
Presented by Dr. John Brunstein: https://www.youtube.com/watch?v=kwx2gezErEg&t=136s
Cannabis varieties are commonly bred or selected for expressed cannabinoid and / or terpenoid profiles. Allelic variation of key genes in the cannabinoid and / or terpenoid pathways would be expected to have phenotypic impact. Thus, an understanding of significant variation in these genes would potentially be useful in underpinning marker assisted selection strategies for breeding novel varieties. To investigate, we have applied the Oxford Nanopore MinION sequencer and the PCR Barcoding coverage. Comparison of these genomic sequences against extant cDNA sequences allows for determination of predicted amino acid sequences for each target. Unlike common short read NGS technologies, where allelic phasing can be unclear, the use of Nanopore long read sequencing may allow for easier resolution of individual parentally derived allelic sequences.
Derived amino acid sequence(s) for each examined gene target were aligned and amino acid substitutions were examined in comparison to paired chemotypic data where available (the majority of samples tested). This approach promises to allow association of particular amino acid substitutions in some target genes with chemotypic effects. Where possible, we have mapped these substitutions back onto available protein crystallographic structures to attempt to assess whether these observed changes are merely linked or are more likely directly mechanistically relevant to chemotype. Overall, we find this method represents a relatively low cost, high yield approach to uncovering markers of utility in directed cannabis breeding programs.
Cannabis dried flower samples were purchased from the BC Liquor Distribution Branch and extracted using KingFisherTM Duo Prime System (Magnetic Particle Processor) and MagMAX Plant DNA Kit in accordance with manufacturer’s instructions.
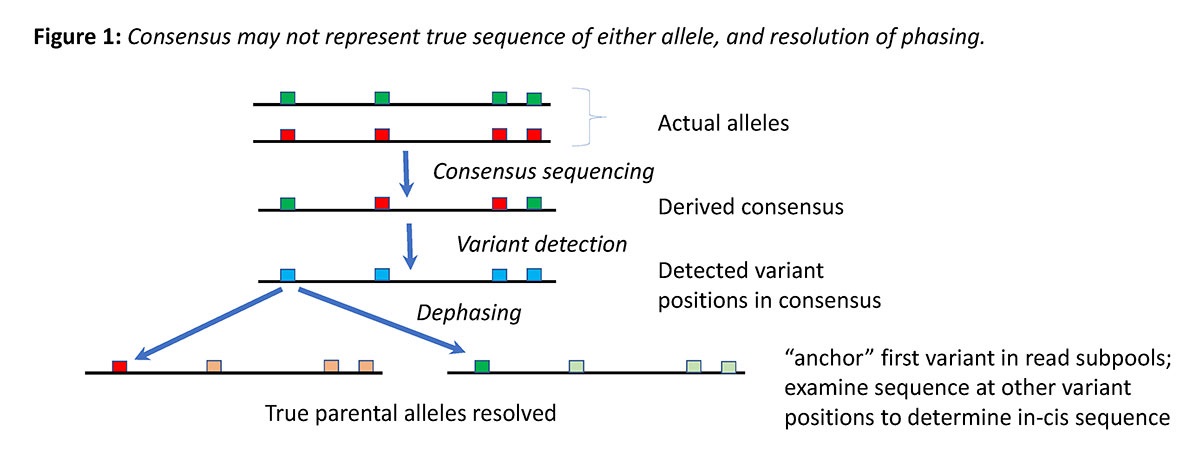
Samples were amplified for individual target genes of interest (see Table 1) using gene-specific primers, developed in house. PCR conditions varied according to expected amplicon size.
For each cannabis sample tested, PCR products for all amplified targets were pooled and barcoded. Up to 12 cannabis samples were then pooled into a single library for NGS. As per Oxford Nanopore Technologies (ONT), the PCR Barcoding Expansion Kit (EXP-PBC001) was used in combination with the Sequencing Ligation Kit (SQK-LSK109) to prepare libraries which were applied to a Flowcell (R9.4.1) and sequenced on the MinION sequencer.
Base calling was performed on ONT MinIT by Guppy. Resulting sequences were demultiplexed by Epi2Me and subsequent analysis steps were performed in CLC Genomics Workbench 10.0.3 (Qiagen).
Chemotypic data (THC(A) and CBD(A) only) were as provided with product lot. For some samples, an expanded panel of analytes including THC(A), CBD(A), plus multiple other cannabinoids and terpenoids were measured by validated in house methods (GC/MS and HPLC).
Barcoded bulk sequence data for each sample was interrogated for target gene sequences using a ‘Map reads to Reference’ function against bait sequences in Table 1. Where matching reads were present, this generated a consensus sequence for the target.
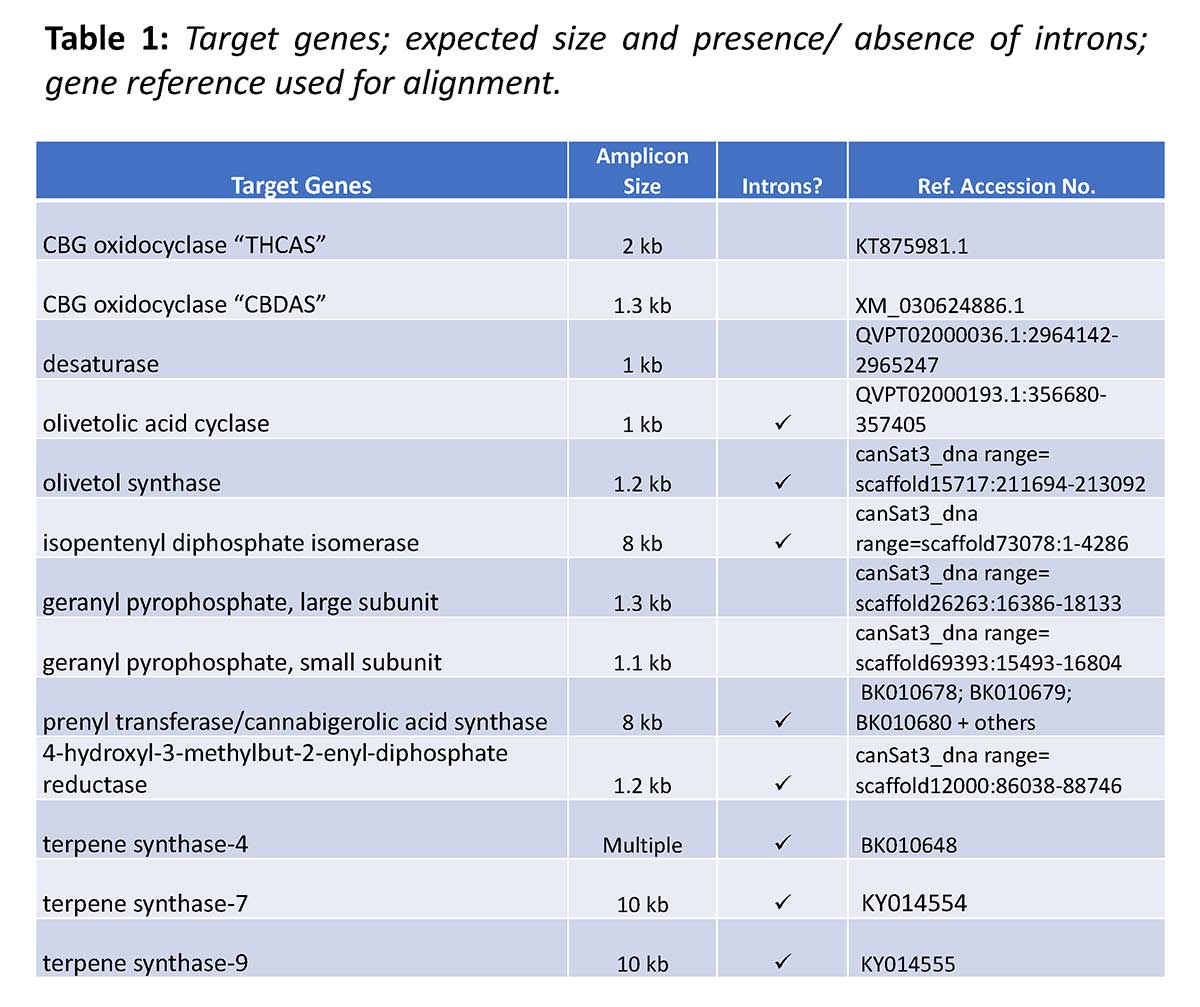
Where more than one copy of a target allele is present, such as commonly the case in diploid organisms, a target consensus sequence such as derived above may in fact represent an artificial mixture of the two true sequences (see Figure 1). This is a problem in both Sanger and widely used short read NGS methods where allelic variation cannot be properly assigned phase (i.e. whether any two heterozygous nucleotide positions are effectively in cis or trans).
To address this, we have attempted to leverage ONT's long read technology to dephase alleles by:
This results in two dephased alleles as with long read technology, variants in common to each allele (i.e. in cis) will statistically associate together in reads grouped this way. We observed heterozygous positions in consensus reads (with near 50:50 ratios) resolve to 90% or better single nucleotide identity in the individual dephased alleles resulting from this method, in line with expected Nanopore accuracy limits.
Where more than two alleles of a locus are present, such as multiple gene copies, sequential iterative rounds of this approach as applied to large enough read depth data sets should effectively resolve out both true sequence of all alleles present, and the copy number. Application of variant finding algorithms on each derived sequence from this method is employed to determine when a unique allele, as opposed to mixed consensus requiring further rounds, is obtained.
For simplicity, in this study we have selected samples where this approach supported evidence of only one or two resolved alleles as noted.
Example: Application to CBG oxidocyclase (CBGO) loci from selected samples

As we cannot rule out undetected alleles or gene loci as a source of cannabinoids present, we are limited to a negative argument that:





COPYRIGHT 2024

